




بیماری اسامای (SMA) یک بیماری آتوزوم مغلوب است که ضعف نورونهای حرکتی تحتانی را تحت تاثیر قرار میدهد. این بیماری در نتیجه جهش ژنتیکی و یا ازدواج خانوادگی احتمال زیادی دارد که بروز کند.
آتروفی عضلانی نخاعی (SMA) گروهی از اختلالات آتوزومی مغلوب را شامل میشود که ضعف رو به رشدِ نورونهای حرکتی تحتانی (LMN) از ویژگیهای آن است. در اوایل دهه 1890 وردنیگ (Werdnig) و هافمن (Hoffman) یک اختلال رو به رشد ماهیچهای را شرح دادند که در نخستین دوره رشد آغاز میشد و نهایتا منجر به مرگ بیماران میشد. مرگ ناشی از این اختلال در ردههای سنی مختلف گزارش شد. از نظر پاتولوژیک، از دست دادن سلولهای ناحیه شاخ پیشین نخاع (Anterior Horn) مشخصه این بیماری است.

نقش کلیدی از دست رفتن نورونهای حرکتی تحتانی در این بیماری، در مطالعات پاتولوژیک متعاقب تایید شد. مطالعاتی که کاهش سلولهای شاخ پیشین نخاع در نخاع و هسته عصبی جمجمهای را نشان میداد. از آن زمان به بعد چندین نوع آتروفی عضلانی نخاعی بر اساس سن بروز علائم بالینی تعریف شدند. معمولترین انواع این بیماری نوع “نوزادیِ حاد” (SMA نوع یک یا بیماری وردینگ هافمن)، نوع “نوزادی مزمن” (بیماری اسامای نوع دو) نوع “نوجوانی مزمن” (SMA نوع سه یا بیماری کوگلبرگ والندر) و در نهایت نوع “بالغ” (بیماری اسامای نوع چهار) هستند.
گفتیم که بیماری اسامای یک بیماری ژنتیکی آتوزومی مغلوب است. نقش ژنتیکی انواع یک تا سه این بیماری بر روی کروموزوم 5q11.2-13.3 قرار دارند. اگر به مطالعه بیشتر در این زمینه علاقهمند هستید این سه مقاله علمی پژوهشیرا دنبال کنید : لینک یک، لینک دو، لینک سه.
سیستمهای طبقهبندی بسیاری برای بیماری اسامای پیشنهاد شده است. این سیستمها غالبا بر مبنای شاخصهای ژنتیکی، بالینی و وراثتی تقسیمبندیها را انجام میدهند. از مهمترین این سیستمها میتوان به طبقهبندی امری (Emery)، طبقهبندی پیرن (Pearn) و سامانه کنسرسیوم SMA بینالمللی (ISMAC) اشاره کرد. از بین این سه ISMAC بیشتر از بقیه مورد تایید است.
تاریخچه و علاتم بالینی بیماری آتروفی عضلانی نخاعی
تشخیص بیماری اسامای در نتیجه یک تاریخچه بالینی دور و دراز بوده است. در صورتی که مبتلایان به بیماری یک تاریخچه کامل خانوادگی ارائه کنند، روند مشاوره ژنتیک تسهیل خواهد شد. بیماران مبتلا به آتروفی عضلانی نخاعی با ضعف و تحلیل عضلانی در دستها و پاها، اندامهای تنفسی و ماهیچههای مغزی دست و پنجه نرم میکنند. این بیماران هیچگونه شواهدی از ناکارامدی مغزی یا دستگاه عصبی مرکزی ندارند. اغلب بیماران مبتلا به آتروفی عضلانی نخاعی از میانگین آیکیو بالایی برخوردار هستند و سطح بالایی از هوشمندی را نشان میدهند.
در ادامه علائم بالینی بیماران مبتلا به انواع مختلف اسامای را شرح داده میشود :
علائم اسامای نوع یک (نوزادی حاد یا بیماری وردینگ هافمن) : بیماران کمتر از 6 ماه سن دارند و 95 درصد از آنها علائم و نشانهها را تا قبل از 3 ماهگی نشان میدهند. آنها با ضعف عضلانی پیشرونده و شدید، سستی و لاغری و هیپوتونی (اختلال در عملکرد ماهیچههای اسکلتی) دست و پنجه نرم میکنند. اخلالات پیاز مغزی (medulla oblongata) به کاهش توانایی خوردن، ضعف در عمل بلع و نارسایی تنفسی منجر میشود. بیماران اسامای نوع یک از نظر ماهیچههای چشمی مشکلی ندارند و ضعف در ماهیچههای صورت نیز عموما اندک و یا اصلا وجود ندارد.
بیشتر بخوانید : تحریم دارویی ایران ؛ تاریخچه، دلایل و تاثیرات تحریم دارو بر کشور
در 30 درصد از موارد نقص حرکتی مرگآفرین مشاهده شده، و 60 درصد از نوزادان مبتلا به SMA نوع یک به سندروم فلاپی نوزاد (FIS) دچار هستند. در هنگام تولد نوزاد ممکن است وجود سیانوز طولانی مدت مشاهده شود. در برخی از موارد این بیماری میتواند به ضعف فولمینانت در نخستین روزهای زندگی بیانجامد. چنین ضعف شدید و اختلال زودهنگام پیاز مغزی امید به زندگی بسیار کوتاهی، با میانگین بقاء 5.9 ماه، به ارمغان میآورد. در 95 درصد از موارد نوزادان در نتیجه عوارض ناشی از بیماری تا 18 ماهگی از بین میروند.
علائم اسامای نوع دو (نوزادی مزمن) : اسامای نوع دو معمولترین فرم آتروفی عضلانی نخاعی است و حتی برخی از متخصصان معتقدند این نوع خود میتواند اساماس نوع یک و سه را نیز در برگیرد. بخش عمدهای از کودکان مبتلا به این بیماری بین 6 تا 18 ماه سن دارند. مهمتریم علائمی که والدین و فیزیسینها به آن اشاره میکنند، تاخیر حرکتی پیشرونده است. نوزدان مبتلا به اسامای نوع دو، عمدتا در سن یکسالگی برای نشستن و یا برخاستن بدون کمک دیگر با مشکلاتی مواجه هستند.

یک ویژگی غیر معمول این بیماری لرزش وضعی بدن به ویژه در انگشتان است. به نظر میرسد این موضوع به انقباض خود به خودی عضلات اسکلتی مرتبط باشد. هایپرتروفی کاذب (افزایش حجم بافت یا اندام در اثر بزرگ شدن سلولها) ماهیچه دوقلو یا گاستروکنمیوس، دفرمه شدن ماهیچههای اسکلتی و نارسایی تنفسی از دیگر عوارضی است که میتواند اتفاق بیافتتد.
طول عمر بیماران مبتلا به اسامای نوع دو از 2 سال تا 3 دهه متغیر است. بخش عمدهای از مرگ و میر در نتیجه این نوع اختلال به خاطر عفونتهای تنفسی است.
علائم اسامای نوع سه (اطفال مزمن) : SMA اطفالِ مزمن نوع خفیف بیماری آتروفی عضلانی نخاعی آتوزوم مغلوب است. این اختلال پس از سن 18 ماهگی در فرد ظاهر میشود. ضعف پروگزیمال پیشرونده آهسته مهمترین مشخه اسامای نوع سه است. بیشتر کودکان مبتلا به این بیماری در نشستن و برخاستن مشکلی ندارند، اما در مهارتهای حرکتی مثل بالا و پایین رفتن از پلهها مشکل دارند.
در اواخر دوران بیماری اختلال پیاز مغزی دیده میشود. مانند بیماران مبتلا به نوع دوم اسامای این افراد نیز ممکن است علائم هایپرتروفی کاذب را نشان دهند. این بیماری به آهستگی پیش میرود و دوره کامل آن خفیف است. بسیاری از بیماران امید به زندگی نرمالی دارند.
علائم اسامای نوع چهار (نوع بزرگسالان) : زمان شروع بیماری به طور معمول در اواسط دهه سوم زندگی است. در بسیاری از موارد در مبتلایان به اسامای نوع چهار علائم مربوط به نوع سوم را مشاهده میکنیم. به طور کلی دوره این بیماری خوشخیم است و افراد از نظر امید به زندگی در وضعیت نرمالی به سر میبرند.
پاتوفیزیولوژی بیماری اساماس (SMA)
در سال 1955، ژن عامل بیماری آتروفی عضلانی نخاعی یا همان بیماری اسامای که به حیات نورون حرکتی آن منجر می شود، به نام ژن SMN مورد شناسایی قرار گرفت. در بدن هر فرد دو ژن SMN1 و SMN2 وجود دارد. بیشتر از 95 درصد از بیماران مبتلا به SMA یک اختلال هوموزیگوت در ژن SMN1 دارند. ژنی که بر روی کروموزوم 5q قرار قرار گرفته است. این اختلال میتوان از نوع جهش، حذف یا بازآرایی (تغییر موقعیت نوکلئوتیدها در رشته دیانای) باشد.
با این حال همه بیماران مبتلا به آتروفی عضلای نخاعی حداقل یک نسخه از SMN2 را حفظ میکنند که در مقایسه با SMN2 تنها 10 درصد از طول پروتئین مربوط به نورون حرکتی (SMN) را تولید میکنند. از این سازمانیافتگی ژنومی میتوان برای توسعه یک مسیر برای درمان بیماری اسامای استفاده کرد. به این صورت که از عملکرد ژن SMN2 که در همه بیماران وجود دارد، برای جبرانِ فقدان ژن SMN1 استفاده کرد.
همهگیرشناسی (اپیدمیولوژی) بیماری SMA
در ایالات متحده آمریکا آتروفی عضلانی نخاعی، پس از فیبروز سیستیک دومین اختلال وراثتی آتوزومی مغلوب به شمار میرود. از هر 10 هزار نوزادی که در این کشور به دنیا میآیند یک نفر به نوع نوزادیِ حاد بیماری اسامای (تیپ یک) مبتلاست. در خصوص فرمهای مزمن (یعنی تیپهای دو و سه) از هر 24 هزار نوزاد متولد شده یک ابتلا به اسامای مشاهده میشود.
از میان تمام بیماران مبتلا به اسامای موجود در ایالات متحده، سهم تیپهای یک و سه این بیماری یک چهارم از کل مبتلایان و یک دوم بقیه نیز به طیف عظیمی از مبتلایانه به SMA تیپ دو تعلق دارد.در مقیاس وسیعتر در سراسر جهان نیز از هر 10 هزار نوزاد تازه متولد شده یک نفر به آتروفی عضلانی نخاعی مبتلاست. از نظر تکرارپذیری نیز از هر 50 نفر یک بیمار مبتلا به SMA را شاهد هستیم.
نرخ مرگ و میر و شیوع بیماری اسامای
نرخ مرگ و میر و نرخ شیوع بیماری اسامای متغیری است که با سن شروع ابتلا به بیماری همبستگی معکوس دارد. در نخستین زمانهای شروع بیماری، بیشترین میزان نزخ مرگ و میر را شاهد هستیم. در بیماران مبتلا به تیپ یک اسامای میانه (M) مدت زمان بقا 7 ماه است. به این گونه که نرخ مرگ و میر 95 درصدی را در ماه 18ام شاهد هستیم. در خصوص مرگ ناشی از اساماس دو نکته حائز همیت است :
- بیشترین میزان مرگ و میر در نتیجه عفونتهای تنفسی اتقاق میافتد.
- در بیماران مبتلا به SMA تیپ دو، سن مرگ گوناگون است، اما مرگ عمدتا در نتیجه عوارض تنفسی اتفاق میافتد.
از نظر جنسیت بیشترین فراوانی مبتلایان به مردان اختصاص دارد. به ویژه در خصوص تیپهای یک و دو که در سنین پایین خودنمایی میکنند. از نظر سن باید سیستم طبقهبندی ISMAC که منحصرا به این موضوع برداخته است، رجوع کرد. بر اساس این سامانه طبقهبندی سن شروع انواع مختلف بیماری آتروفی عضلانی نخاعی به قرار زیر است :
- اسامای نوع یک (نوزادی حاد یا بیماری وردینگ هافمن) : سن شروع بیماری از تولد تا 6 ماهگی است.
- اسامای نوع دو (نوزادی مزمن) : سن شروع بیماری از 6 تا 18 ماهگی است.
- اسامای نوع سه (اطفال مزمن) : سن شروع بیماری از 18 ماهگی به بعد است.
- اسامای نوع چهار (نوع بزرگسالان) : سن شروع بیماری در بزرگسالی و به طور میانگین در دهه سوم زندگی است.
وضعیت فیزیکی بیماران SMA چگونه است؟
افراد مبتلا به این بیماری مربوط به نورونهای حرکتی تحتانی، دچار ضعف و سستی، هیپوتونی، کمبود یا فقدان رفلکسهای تاندونی عمیق، ضعف در انقباض خود به خودی و در نهایت آتروفی عضلانی هستند.
در بیماران اسامای نوع یک (نوزادی حاد یا بیماری وردینگ هافمن)، شعف عضلانی منتشره و هیپوتونی در کنار انواعی از مانورهای جانبی مانند پاسخهای جانبی، تعلیقهای عمودی و افقی قابل مشاهده است. به طور کلی نوزادان مبتلا به نوع یکِ اسامای، وقتی درحالت نشسته قرار دارند نمیتوانند سر خود را بالا نگه دارند. همچنین وقتی شخصی آنها را به طور عمودی نگه میدارد معمولا دچار لغزش میشوند. هنگامی که پزشک آنها را از زیر شکم و صورت نگه به صورت افقی نگه میدارد، سستی و شل بودن در وضعیت آنها به چشم میخورد.
بیشتر بخوانید : مصرف ماری جوانا موجب کاهش علائم بیماری التهاب روده میشود!
ضعف عضلات پروگزیمال از عضلات جانبی بیشتر است و همچنین ممکن است به بیماری ماهیچهای یا میوپاتی نیز دچار باشند. یافتههای مربوط به معاینات حسی در این بیماران طبیعی است. رفلکسهای تاندونی عمیق در مبتلایان به اسامای نوع یک دیده نمیشود. انقباضهای طولانی مدت و اختلالات اسفنکتر نیز از دیگر علائم این بیماری است.
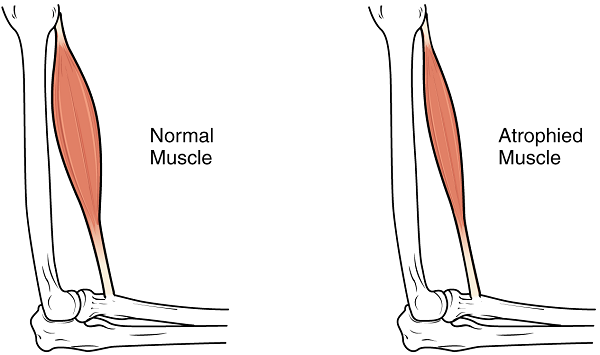
آرتروگریپوز (Arthrogryposis) با دفرمه شدن دستها، پاها و مفاصل در هنگام تولد را در نتیجه هیپوتونی رحمی میتوان مشاهده کرد. اسکلیوزیس یا انحنای غیر طبیعی در ستون فقرات نیز ممکن است در این افراد مشاهده شود. در نوزادان یا افراد تازه متولد شده نقص در انقباضهای خود به خودی اغلب محدود به زبان است، اما تشخیص آن از حرکات تصادفی نرمال بسیار دشوار است، مگر آنکه آتروفی در فرد مشخص باشد.
در بیماران اساماس نوع دو (نوزادی مزمن)، بالاخص نوزدان، توانایی نشستن به صورت مستقل وجود ندارد، هرچند درصورتی که آنها را در آن موقعیت قرار دهیم، ممکن است به صورت مستقیم باقی بمانند. مانند تیپهای یک و دو، ضعف پروگزیمال متقارن، هیپوتونی و نقض در انقباض خود به خودی از جمله علائم قابل ذکر است. مانند مورد قبلی تستهای حسی نرمال است و علائمی از انقباضهای طولانی مدت دیده نمیشود. هنگامی که دستان این بیماران را میگیرید میتوانید لرزشهای موضعی را نیز احساس کنید.
در بیماران مبتلا به اسامای نوع سه (اطفال مزمن یا سندرم کوگلبرگ ولاندر)، کودکان با وجود آنکه میتوانند راه بروند، اما از ضعف ماهیچه پروگزیمال و سطوح مختلفی از هیپوتونی و تحلیل عضلانی رنج میبرند. در این بیماری اندامهای تحتانی بیشتر از اندامهای فوقانی تحت تاثیر قرار میگیرند.
اسامای نوع چهار (نوع بزرگسالان) نیز از نظر علائم بالینی شباهتهای زیادی به نوع سه دارد. با این وجود درجه کلی ضعف حرکتی در افراد مبتلا به اسامای نوع 3 شدت بیشتری دارد.
دلیل ابتلا به بیماری اسامای چیست؟
همانطور که بخش پاتوفیزیولوژی اشاره کردیم، در سال 1995، ژن SMN که مسئول بروز تیپهای یک تا سه آتروفی عضلانی نخاعی است، در بازوی بلند کروموزوم 5 مورد شناسایی قرار گرفت. در بازوی 5q دو نسخه از ژن SMN مورد شناسایی قرار گرفته است. نوع اول نوع تلومر (telomeric) یا SMNt و دیگری نوع سنترومر (centromeric) یا SMNc است. بجز تغییراتی در جفت بازهای آلی در اگزونهای 7 و 8 تفاوت دیگری میان این دو ژن دیده نمیشود. حدود 95 درصد از موارد بیماری SMA به حذف یک هموزیگوت در ژن SMNt مربوط است.
بیان ژن SMN1 پروتئینی به نام SMN را بوجود میآورد. در نقطه مقابل، بیان ژن SMN2 یک نسخه کوتاهتر از همان پروتئین را تولید میکند که 16 آمینواسید که فاقد 16 آمینو اسید در سیترمنیال (C-termenal) است. جابهجایی یک جفت باز آلی در اگزون شماره 7 ژن SMN2 منجر به شکلگیری این پروتئین کوتاه شده میشود. این جابهجایی منجر میشود mRNA حاصل در محلی تاب بخورد، در نتیجه توالی اگزون شماره 7 به طور کامل حذف شود. حدود 70 تا 80 درصد از محصول این ژن به شکل همین پروتئین کوتاهشده است. تنها حدود 10 تا 25 درصد از پروتئینهای ترجمه شده از mRNA ژن SMN از نظر طولانی کامل هستند.
حذف یا جهش در ژن SMN1 به طور قابل ملاحظهای منجر به کاهش بیان پروتئین SMN میشود. بیان ژن SMN2 به تنهایی نمیتواند مقدار کافی پروتئین تولید کند تا در نورونهای حرکتی تحتانی عمل پردازش mRNA به طور نرمال صورت گیرد. همبستگی میان تعداد نسخههای ژن SMN2 و فنوتیپ این بیماری اینگونه است که هرچه تعداد نسخههای بیشتر باشد بیماری خفیفتر خواهد بود.

به علاوه سطح پایین پروتئین SMN فرمهای شدیدی از این بیماری را به دنبال خود میآورد. پردازش ناکارآمد و غیرطبیعی mRNA منجر به سمی شدن نورونهای حرکتی تحتانی و از بین رفتن سلولها میشود. پروتئین SMN بخشی از یک پروتئین مالتیمریک است که نقشی حیاتی در تجمیع snRNPها دارد. {ویکی: اینها مولکولهای کوچکی هستند که از پروتئین و رنا ساخته شدهاند} snRNPها در تشکیل حلقه در پیش mRNAها ضروری هستند و اثر سمی بر عملکرد سلولی طبیعی دارند.
اگرچ پروتئین SMN در بسیاری از انواع نورونها و اندامهای بدن بیان میشود، اما دلیل اینکه این جهش به نابودی انتخابی نورونهای حرکتی تحتانی منتج میشود نامشخص است. ژنِ پروتئین مهارکننده آپوپتوز عصبی (NAIP) نیز در سال 1955 مورد شناسایی قرار گرفت. حذف این ژنها به صورت هموزیگوت در 45 درصد از بیماران مبتلا به بیماری اسامای نوع یک و 18 درصد از مبتلایان به نوع دو و سه مشاهده شده است. این ژن به رده پروتئینهای مهارکننده آپوپتوزِ (AIP) بسیار حفاظت شده تعلق دارد که به تنظیم مرگ برنامهریزی شده سلول کمک میکند. مشاهدات نشان دادهاند که حذف این ژن منجر به وخیم شدن فنوتیپ بیماری اسامای میشود.
مراقبت و درمان بیماری اسامای (SMA)
در دسامبر 2016، سازمان غذا و داروی ایالات متحده (FDA) دارویی تحت عنوان نوزینرسن (nusinersen) یا با نام معروفتر اسپینرازا (Spinraza) به عنوان نخستین داروی برای درمان اسامای در کودکان و بزرگسالان تایید کرد. نوزینرسن یک اولیگونوکلئوتید آنتیسنس (ASO) است که برای درمان بیماری اسامای به کار گرفته میشود. بیماریای که در آن جهش در کروموزوم 5q به ناکارآمدی پروتئین SMN منتج میشود.
با استفاده از ارزیابیهای آزمایشگاهی و مدلهای حیوانات تراریخته مبتلا به اسامای و تاثیر اسپینرازا بر آن نشان داده شد که این دارو منجر به افزایش بازدهی اگزون شماره 7 در تولید رونوشت mRNA ژن SMN2 و تولید پروتئین کامل میشود.
در خصوص دستگاه تنفسی که شایعترین عمال مرگ و میر در انواع یک و دو اسامای است، به دلیل فقدان تحریک عصبی مناسب، ماهیچههای میان دندهای ضعف دارند. به علاوه دیافراگم نیز دیگر کارایی خود را ندارد. در نتیجه به دلیل ضعف این عضلات انجام تنفس و اعمال مانند سرفه با مشکل مواجه خواهد شد. مشکل در تنفس همانا و عدم اکسیژنرسانی کافی به اندامها همانا. از اینها گذشته ترشحهای موکوزی مجاری تنفسی نیز به خوبی دفع نمیشود. موضوعی که در هنگام خواب بیشتر میشود. در نتیجه این احتمال تقویت میشود که ترشحاتی نظیر بزاق و خلط به درون مجاری تنفسی وارد و آسپیراسیون به بار آید. سرفههای ضعیف از بازگشت این ترشحات جلوگیری میکند و درنتیجه عفونتهای تنفسی و در نهایت مرگ درانتظار بیمار نشسته است.
بیشتر بخوانید : بیماری اعتیاد به بازی های دیجیتالی به رسمیت شناخته میشود!
برای درمان این معضل و خارج کردن ترشحات ریوی چه به صورت دستی و چه به صورت مکانیکی میبایست فیزیوتراپی بر روی بیمار انجام شود. در این وضعیت برای بهبود فرآیند تنفس بیماران تهویه غیرتهاجمی تنفسی یا BiPAP انجام میگیرد و اگر بیماری خیلی حاد باشد، نایشکافی یا تراکستومی بر روی بیمار انجام میشود. تراکستومی تکلم بیمار را مختل میکند ولی در مجموع به طور قابل ملاحظهای طول عمر فرد متبلا را افزایش میدهد.
افراد مبتلا به اسامای با مشکلات تغذیهای مواجه هستند. ناتوانی در بازکردن دهان، بلعیدن غذا و نیز جویدن از جمله اختلالات آنهاست. این موضوع خطر بسیار جدی برای نوزادان به شمار میرود و رشد آنها را تهدید میکند. در برخی از موارد، همراه با عدم تحرک بیمار، مواد غذایی در موعد مقرر خود از معده عبور نمیکنند. همچنین بازگشت اسید معده به مری، یبوست، استفراغ و نفخ از دیگر عوارض مرتبط با دستگاه گوارش این بیماران است. در موارد حاد نوع یک و دو، با استفاده از عمل جراحی لوله تغذیه معدی کار گذاشته میشود.
این بیماری متابولیسم بدن را نیز تهدید میکند. در نتیجه ناکارآمدی بتا اکسیداسیون اسیدهای چرب، ماهیچهها مختل شده و ارگانیک اسیدمی به بار میآید. به ویژه اگر بیمار به مدت طولانی گرسنگیرا تجربه کرده باشد. به همین دلیل یکی از توصیههای که به بیمار اسامای می شود پرهیز از مصرف چربیها و گرسنگی طولانی مدت است. پرهیز از مصرف غذاهایی که به سختی هضم میشود نیز مهم است.
در اثر ضعف ماهیچهای مشکلات اسکلتی متعددی مانند سفت شدن مفاصل، دررفتگی لگن، انحراف ستون مهره، درد و پوکی استخوان و افزایش احتمال شکستگی استخوان در بیماران اسامای ایجاد میشود. در انواع یک و دو بیماری اسامای، در سنین 8 تا 10 سالگی، برای کاهش فشار بر روی ستون فقرات (که در نتیجه ضعف عضلانی دچار ثبات مفصلی، انقباض موضعی و گوژپشتی شدهاند) و نیز بهبود فرآیند تنفس، جراحی “جوش دادن مهرهها” انجام میشود. در کنار جراحی، نرمشهای حرکتی، تقویت استخوانها و استفاده از دستگاههای تغییر وضعیت بدن میتواند به طور قابل توجهای عوارض جانبی بیماری را کاهش دهد.

در زمینه حرکتهای روزمره نیز این بیماران به حمایتهای حرکتی نیاز دارند و ارتزها (orthosis) مانند ارتز مچ پا یا ارتزهای سینهای-کمری در حفظ و تثبیت وضعیت و حرکت کردن آنها بسیار موثر است. این ابزارها کیفیت زندگی این بیماران را به طرز قابل ملاحظهای افزایش میدهد.
نکته قابل ملاجظه این است که بر اساس مشاهدات و گزارشها کودکانی که به آتروف عضلانی نخاعی دچار هستند از لحاظ رفتاری تفاوت چندانی با افراد سالم ندارند. این افراد حتی تکامل شناختیشان سریعتر رخ داده و برخی از جنبههای هوششان از میانگین جامعه هم بالاتر است. به همین دلیل است که علیرغم ناتوانیهای فیزیکی که دارند، امید و رضایتمندی از زندگیشان در سطح بالاتری نسبت به سایر افراد است.
هزینه درمان بیماری اسامای (SMA) چقدر است؟
باید بر این نکته تاکید کرد که بیماری آتروفی عضلانی نخاعی به عنوان یک بیماری آتوزوم مغلوب عمدتا در فرزندان حاصل از ازدواجهای فامیلی رخ میدهد. تصور کنید فردی مبتلا به اسامای (با آلل فرضی aa) با فرد سالمی ازدواج میکند. در این صورت در همه نسلهای بعدی وی آلل بیماریزایی (a) حداقل به صورت ناقل وجود خواهد داشت. به همین دلیل در ازدواجهای فامیلی احتمال تولد فرزندی با فنوتیپ مغلوب بیشتر از سایر انواع ازدواجها وجود دارد.
در این ازدواجهای فامیلی بسیار رایج و در نتیجه احتمال بروز این صفت بیشتر است. هرچند متاسفانه در این زمینه آماری در دست نیست. به دلیل عدم وجود اطلاعرسانیهای لازم، این بیماری برعکس سندرم داون در غربالگریهای پیش از بارداری قرار ندارد. اگر چنین میبود، احتمالا امروز تعداد مبتلایان به آن بسیار کمتر از این تعداد میبود. تفکر نادرستی که در میان مردم وجود دارد این است که این بیماری یک بیماری نادر است و درصد کمی از مردم به آن مبتلا هستند.
در میان یه تیپ عمده بیماری اسامای هرچه از تیپ یک به سوی تیپ سه حرکت میکنیم، طول عمر افراد بیشتر و بیشتر میشود. بالاتر نیز به تفضیل توضیح دادیم که بیماران تیپ یک، در همان دوران نوزادی عمدتا از بین میروند، در حالی که مبتلایان به تیپ دو تا چهار یا پنج سالگی و بیماران تیپ سه حتی تا بعدها پس از سن بلوغ نیز به زندگی ادامه میدهند.
یک تفاوت عمده که میان بیماری اسامای با بیماریهای کشنده دیگر نظیر سرطان و غیره وجود دارد این است که عامل بیماری به هیچ یک از ارگانهای فرد بیمار حمله نمیکند. با این وجود در بسیاری از موارد عمل جراحی تراکستومی یا نایبری انجام میشود و بدین طریق عملا زندگی نباتی بیمار آغاز میشود. نقدی که امروزه در خصوص نحوه برخورد بیمارستانهای با این بیماران وارد است به این نکته اشاره میکند که بیمارستانها برای ترخیص زودتر کودک اسامای به عمل جراحی تراکستومی روی میآورد.
برخی از بیماران و خانوادههایشان راضی به این کار نمیشوند و ترجیح میدهند با بیمار خود را اینتوپه و ایکستوپه کنند. اینتوپه به عملی گفته میشود که با سوراخ کردن گلو لولهای در ریه کودک قرار میگیرد و اکستوبه هم به این معنی است که ماسک روی صورت میگذارند.
اسپینرازا 7.5 میلیارد تومانی تنها داروی مورد تایید
پیشتر نیز اشاره کردیم که داروی اسپینرازا که در سال 2016 توسط سازمان غذا و داروی ایالات متحده (FDA) برای درمان SMA مورد استفاده قرار میگیرد. هزینه این دارو در ایران برای یک دوره یکساله و شش آمپول 750 هزار دلار است (با احتساب دلار 10 هزار تومانی 7.5 میلیارد تومان). به نقل از خبرآنلاین والدین یکی از کودکان مبتلا در این زمینه میگویند :
این دارو قبلا به صورت طرح تحقیقاتی بود که اگر مسئولان و پزشکان ما تلاش میکردند، میتوانستند به صورت طرح تحقیقاتی این دارو را رایگان به ایران بیاورند و در صورت حصول نتیجه مثبت بیمه آن را تایید میکرد. اگر این اتفاق میافتاد، چه بسا بسیاری از کودکان SMA درد نمیکشیدند و زنده میماندند.
این هزینه قیمت ارائه شده در آمریکا است و اگر مسئولان تلاش میکردند و شرکت داروسازی بدون کسب سود با آنها رو به رو میشد، ایران میتوانست پای مذاکره برود و با قیمت بسیار کمتری این دارو را وارد کند.
چرا که قیمت این دارو به صورت منطقهای تعیین میشود. از سوی دیگر، مسئولان میگویند هیچشرکت داروسازی اعلام آمادگی نکرده است، در حالی که شرکتهای داروسازی پشتوانه مالی ندارند. چگونه خانواده یک کودک با بیماری SMA توانایی پرداخت این مبلغ را دارد؟ ما در حال حاضر در تلاش هستیم که برای کمک به بیماران SMA از طریق کمپین پول جمع کنیم.
آمپول اسپینزارا به مدت 6 مرتبه در یک بازه زمانی یکساله در نخاع تزریق میشود. یکی دیگر از نقدهای وارده این است که وزارت بهداشت میبایست اسپینرازا ر در فهرست داروهای مورد تایید قرار دهد و برای آن بودجه اختصاص دهد. مادر رادین، یکی از کودکان مبتلا به این بیماری میگوید :
نمیتوان گفت که درمان با اسپینرازا قطعی است، ولی تنها داروی مورد تایید جهانی است. از سوی دیگر، داروهای دیگری در مرحله تحقیقاتی قرار دارند که اگر مسئولان تلاش میکردند میتوانستند این داروها را به ایران بیاورند. یکی از این داروها روژه نام دارد که داروی خوراکی است و دیگری ژن درمانی است که با تزریق ژن معیوب اصلاح میشود، اما دوباره ما شاهد کم کاری مسئولان در این باره هستیم.
داروی اسپینرازا در اواخر 2016 و اوایل 2017 توانست تائیدیه سازمان غذا و داروی ایالات متحده (FDA) را کسب کند. این دارو با نام ژنریک Nusinersen به صورت ویالهای 12 و 5 میلیگرمی توسط کمپانی بایوژن (BioGene) در آمریکا تولید میشود. اسپینرازا به صورت مستقیم به درون مایع مغزی نخاعی تزریق میشود. در شروع درمان سه دوز به به فاصله ۱۴ روز و دوز چهارم با فاصله ۳۰ روز تزریق نخاعی می گردد و در ادامه هر ۴ ماه یک دوز مصرف می شود.
بیشتر بخوانید :
- روان پریشی مشترک چیست ؛ آیا توهمات روانی مانند بیماریهای واگیردار به اعضای خانواده منتقل میشود؟
- نخستین درمانهای انسانی کریسپر در سال ۲۰۱۸ ؛ پایانی برای بیماریهای لاعلاج!
- عجیبترین بیماریهای روانی جهان ؛ وقتی مرزی بین واقعیت و خیال وجود ندارد!
- بیماری ایدز چیست؟ در مورد اچ آی وی بیشتر بدانیم!
منبع : MedScape